INTRODUCTION
The oral route of drug administration is the most versatile, convenient, and often employed route. However, fluctuation in drug concentration in the blood stream and tissues with the resulting toxicity are some of the shortcomings associated with conventional oral tablets. Frequent drug administration vis-à-vis drug adherence are other problems associated with conventional dosage forms.1,2,3 To obviate these shortcomings, controlled release formulations, especially those for oral administration, have been investigated and developed with the sole aim of maintaining a constant drug concentration in the blood stream for longer through slow release of drug into the gastrointestinal tract (GIT).4 Although the oral route is the most preferred for drug administration, studies has demonstrated two physiological influences: short gastric residence time and variable gastric emptying time. Thus, bioavailability and time to achieve maximum plasma concentration cannot be predicted. It must be noted that most drugs are absorbed in the stomach and upper part of the intestine. However, residence time within these regions is short (2 to 3 h). Hence any methods to prolong the residence time of drugs within these regions will improve bioavailability and therapeutic outcome.4,5
The oral route has received greater attention and given more successful outcomes than any other route in controlled drug delivery systems.6,7 This is not unconnected with the physiology of the GIT, which offers more flexibility in the design of oral dosage forms compared with other routes.8,9,10 The most crucial challenge with an oral controlled drug delivery device is not just sustaining the drug release, but also ensuring that the dosage form is sufficiently prolonged within the GIT for complete release from the device. Scientists and the pharmaceutical industries, right from the first generation of controlled release (1952 to the 1970s) to the second generation (1980 to 2010), have made major breakthroughs in the development of oral controlled drug delivery systems by working against gastrointestinal emptying.11
One such device employs the concept of the gastroretentive drug delivery system (GRDDS).12,13 Oral dosage forms for the GRDDS have received much attention over the years for enabling control over the time and site of drug release.2,12 Prolongation of the gastric retention of drug delivery devices has numerous advantages. These include better absorption, enhanced bioavailability and therapeutic efficacy, and possible reduction of dose size.14
The major principle of the GRDDS is prolongation of stay of the dosage form and the release of drug at the absorption site. Many approaches have been adopted, but the most recent is “the floating device”.15 Floating dosage forms have low bulk density, hence their ability to float in the gastric fluid for a long time, thus contributing to improved bioavailability.16 A floating device can also be improved upon by incorporating a combination of two or more active pharmaceutical ingredients (APIs) in a single dosage form (multilayer tablets). Multilayer tablets can be used to obviate chemical incompatibilities between APIs through physical separation and also to achieve different drug release profiles, e.g., immediate release and extended release segments.17 Such an approach can be used for the formulation of sustained release tablets comprising an immediate release outer layer and a maintenance inner layer. This has been employed to overcome single-layered tablets’ fluctuation in drug concentration both in the blood stream and at the site of action.18,19 Drugs that are mainly absorbed from the upper part of the GIT, such as albuterol, furosemide, and theophylline, are worthy candidates. Development of these drugs in floating sustained release dosage form helps to prolong their limited bioavailability.20
Theophylline has an antiinflammatory property at the therapeutic regular dose and as such plays an important role in treating chronic obstructive pulmonary disease.21 Theophylline has a narrow therapeutic index (10-20 µg/mL); thus the conventional preparations experience fluctuation between maximum and minimum blood concentration, resulting in poor therapeutic outcome. On the other hand, patients on regular sustained release preparations may experience delay in the onset of drug action since the initial release may not be therapeutic. Thus, in the current study, bilayer theophylline matrix tablets were formulated by double compression using granules produced by polymeric granulation and simple coacervation. One layer provides the immediate release component, while the second layer provides the sustained release segment.
MATERIALS AND METHODS
Materials
The test drug (theophylline powder) was obtained from Vital Biotic, Nigeria Ltd. as a free sample.
Excipients and reagents
Absolute ethanol, citric acid, and sodium bicarbonate (Guangdong Guanghua Sci-Tech Co. Ltd., Shantou, Guangdong, China); carboxymethyl cellulose (CMC) and lactose (Kermel); and normal saline (Unique Pharmaceutical Nigeria Ltd.) were obtained. Acrylic-methacrylic polymer (Eudragit RL100) was received as a gift sample from Evonik Industries AG-Werk Röhm, Darmstadt, Germany. Amaranth solution (Vinayak Ingredients Pvt Ltd, India) and magnesium stearate, talc, and maize starch (Kermel) were also used.
Ethical approval
No ethical approval is required by the Delta State University for research of this nature since the work does not involve animal studies or clinical trials; however, theophylline is a controlled drug in some countries, hence the need for ethical approval. The research work was approved by the Faculty of Basic Medical Sciences Research and Ethics Committee of the Delta State University, Abraka, Nigeria. The approval number is REC/FBMS/DELSU/19/45.
Methods
To formulate bilayer floating theophylline tablets, two sets of granules (conventional granules for the immediate release segment and a second set of granules for the prolonged release segment) were formulated.
Granules for the immediate release layer (IRL)
To form the immediate release section, conventional granules were formulated by wet granulation (F6, Table 1). A 1.5 g sample of maize starch powder was weighed and converted to mucilage with boiled water. Theophylline powder (25 g), lactose (18.5 g), and maize starch powder (2 g) were weighed and transferred into a clean porcelain mortar. A few drops of amaranth solution (colorant) were added and thoroughly blended. The blend was kneaded with maize starch mucilage to form a damp mass. This mass was forced through a 1 mm sieve and dried with a hot air oven (Lead Engineering, St Helens, UK. Model: GP/50/CLAD/100/HYD) at 60±0.5°C for 24 h. The dried mass was passed through a sieve (710 µm) and characterized by measuring the flow and packing property before storage.
Preparation of granules for the sustained release layer (SRL)
Granules for the SRL were prepared according to the formula in Table 1. A 5 g sample of Eudragit RL100 (10% w/w) was weighed and dissolved in 30 mL of absolute ethanol. A sample of theophylline powder (20 g) and a 10 g sample of CMC were weighed and transferred to a clean mortar and thoroughly blended. The powder blends were kneaded with the Eudragit-ethanol mixture to form a wet mass. The wet mass was forced through a 1 mm sieve and dried in a hot air oven at 50.0±1.0°C for 2 h. The dried granules were passed through a 710 µm sieve to form the required granules (batch F1, Table 1). In other experiments, the quantities of Eudragit and CMC were varied in order to form batches F2, F3, and F5.
Batch F4 was prepared by a simple coacervation technique. Here, a 10 g sample (20% w/w) of Eudragit RL100 was weighed and dissolved in 150 mL of absolute ethanol. A 20 g sample of theophylline powder was weighed and mixed with the Eudragit-ethanol mixture. Thereafter, 350 mL of normal saline solution (0.9% w/v of sodium chloride) was added followed by stirring to form coacervates. The coacervates so formed were allowed to settle, filtered, and dried in a hot air oven at 50.0±1.0 oC for 2 h. The dried mass was passed through a 710-µm sieve to form the required granules. The granules were characterized before storage for further study.
Preformulation studies
a- Angle of repose: A 20 g sample of granules was weighed and allowed to flow through a funnel orifice at a height of 7 cm. The height and diameter of the cone so formed were measured. The procedure was performed in triplicate and the mean value recorded. The angle of repose (θ) was computed from equation (1):
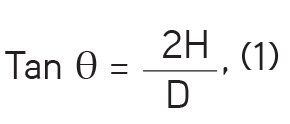
where H and D are the height and diameter of the powder cone so formed.
b- Densities and compressibility index (CI): A sample of granules (20 g) was weighed and transferred into a 100-mL cylinder of an automated tapped density tester (Model C-TDA2, Campbell Electronics, Mumbai, India). The volumeter was allowed to tap 100 times and the tapped volume recorded. The bulk and tapped densities and the CI were computed from equations (2), (3), and (4). The procedure was performed in triplicate and the mean values recorded.

c- Particle size analysis: A sample of granules (50 g) was weighed and transferred into the topmost sieve of a set of sieves arranged in descending order. The set of sieves was shaken with a sieve shaker (Endecott Ltd, UK) for 5 min. The quantity of granules in each sieve was weighed to determine the size distribution. The procedure was performed in triplicate and mean values recorded.
Compression of granules to tablets
A sample of granules (500 mg) for the SRL was weighed and poured into the die cavity of a single punch tableting machine (Kilian & Co GMBH Kolu-Niel, Type KS 043111-196, Buchschlag, Germany). A 100 mg sample of granules for the IRL was weighed and transferred to the same die cavity. This was compressed into bilayer tablets at a force of 28 kgF without agitation. The compression force was kept constant and the procedure repeated for all the batches.
Evaluation of tablets
i- Percentage weight variability: Twenty tablets were selected at random and the mean weight of each was determined with the aid of an analytical balance (Shimadzu Philippines Manufacturing Inc.). The percentage weight variability was computed using equation (6):
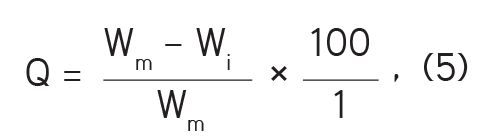
where wm is the mean weight and wi is the weight of each tablet.
ii- Tablets’ tensile strength determination: The diameter (d), thickness (t), and crushing load (P) of each 10 tablets selected at random were determined using a Veego digital hardness test apparatus. The mean tensile strength of the tablets was determined using equation (6):
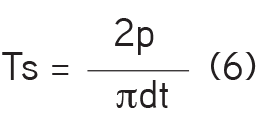
iii- Disintegration test: The method described in the British Pharmacopoeia22 was employed. Six tablets were selected at random from each batch and a tablet was placed in each of the six baskets of the disintegration apparatus (Manesty Machine, MK4, UK). The baskets were immersed in warm distilled water maintained at 37±1°C. The mean time taken for the tablets to break up and pass completely through the mesh was recorded as the disintegration time.
iv- Friability test: To evaluate the degree of friability of the tablets, ten tablets were picked at random and weighed. The tablets were placed in the drum of a friabilator (Erweka friabilator). The machine was operated at 25 rpm for 4 min. The tablets were removed from the friabilator, dedusted, and reweighed. The difference in the initial and final weights expressed as a percentage was recorded as the friability.
v- Dissolution test: This test was carried out using the rotating basket method (USP apparatus one). The dissolution medium was 0.1 N hydrochloric acid (pH 2.3). The apparatus consisted of a Pyrex glass vessel containing 900 mL of the dissolution medium maintained at 37±1°C and a cylindrical basket made of stainless-steel wire mesh (aperture size 425 µm). One tablet was placed in the basket, which was rotated at 100 rpm in the dissolution medium. Aliquots (5 mL) were withdrawn at specified time intervals and the amount of drug released was determined using a ultraviolet (UV) spectrophotometer (PG Instrument, USA) at a wavelength of 272 nm. Fresh dissolution medium (5 mL) was added each time a sample was withdrawn.
Theophylline analysis (calibration curve): To standardize theophylline release from the various formulations, a standard calibration curve of theophylline was prepared as follows. A sample of theophylline powder (100 mg) was weighed with an analytical balance and dissolved in 100 mL of medium (0.1 N hydrochloric acid) to obtain a solution of 1 mg/mL (i.e. dilution X1). A 10 mL sample of X1 was measured and diluted with 0.1 N HCl to 100 mL to obtain a solution of 0.1 mg/mL (X2). This process of serial dilution continued until solutions of 3, 5, 7, 9, 11, 13, 15, and 17 µg/mL were obtained. The absorbances of these standard solutions were measured at a wavelength of 272 nm using a UV spectrophotometer. The tests were conducted in triplicate and mean values recorded. Plots of mean absorbance against concentrations were made and a linear regression coefficient (R2 values) of 0.9947 obtained. The same procedure was used to compute the amount of theophylline released into the dissolution medium at various time intervals.
vi- Kinetic data analysis: Data obtained from the dissolution study were fitted into three well known release models [equations (7), (8), and (9)]:
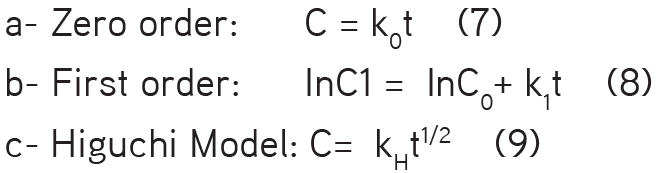
Here C0 is the initial amount of drug in the dosage form, C is the percentage amount of drug released, and C1 is the percentage of residual drug at time t. K0, K1, and KH, are the zero order, first order, and Higuchi constants, respectively.
vii- Buoyancy lag time and floating time: A tablet was selected from each batch at random and placed in a 1000 mL beaker containing 900 mL of 0.1 N HCl maintained at 37±1°C. The time required for the tablet to rise to the surface was recorded as the buoyancy lag time, while the duration of floating on the surface without rupturing was recorded as the total floating time determined by visual observation.
viii- Swelling time: The extent of swelling was measured in terms of percentage weight gained by the tablets. A tablet was selected from each batch, weighed, and kept in a beaker containing 900 mL of 0.1 N HCl solution at 37±1°C. The tablet was withdrawn from the beaker at a specified time interval (swelling time interval is 2 h); then excess HCl was blotted with tissue paper and the tablet weighed. Percentage weight gain by the tablet was computed with equation (10):
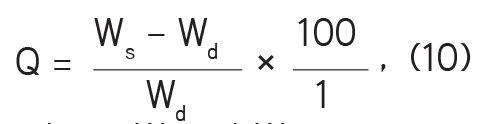
where Ws and Wd represent the weight of the swollen tablet and initial weight before swelling, respectively.
ix- Assay procedure (content uniformity): The theophylline assay of the various batches was performed according to the pharmacopeia method.23 In this method, 2 tablets from each batch were crushed and 375 mg (equivalent to 240 mg of theophylline) was weighed and dissolved in 100 mL of distilled water. A sample (20 mL) of 0.1 M silver nitrate was added and shaken properly for 10 min. The solution so formed was titrated with 0.1 M sodium hydroxide solution using bromothymol blue solution as indicator. Each milliliter of 0.1 M sodium hydroxide solution is equivalent to 18.02 mg of theophylline.
Statistical analysis
All data were expressed as mean ± standard deviation of three determinations. Differences between means were determined with One-Way ANOVA at p<0.05.
RESULTS AND DISCUSSION
Packing and flow properties
The results for the packing and flow properties such as bulk and tapped densities, the Hausner ratio, Carr’s CI, flow rate, and angle of repose are shown in Table 2. The angle of repose for all formulations was within the range of 29.07 ° to 34.46 ° except batch F4 (angle of repose was 40.58 °), which was prepared by simple coacervation technique. Angle of repose is an indication of powder flowability;24 all formulations except batch F4 had good flow. Batch F4 exhibited passable (may hang up, flow aid needed) type of flow. The passable flow of batch F4 may be because most of the particles are below 250 µm in size (Figure 1).
“Particles larger than 250 µm are usually relatively free flowing but as the size falls below 100 µm, powders become cohesive and flow problems are likely to occur”.25
The CI for all granule formulation varied between 6.33% and 10.45% except for batches F4 and F5. Batch F5’s CI value was 12.34% (good flow), while that of batch F4 was 22.15%. These variations could be due to the type and concentrations of the binders used. Combination of Eudragit® RL100 and CMC produced granules with better flow.
Physicochemical properties of the bilayer floating tablets
The physicochemical properties of the various tablets such as hardness, weight variability, friability, and disintegration time are presented in Table 3. The hardness of the tablets in all batches ranged between 4.74 kgF and 9.84 kgF. The hardness value of the batch that contained only CMC (batch F5) was 6.08 kgF, while batches F1, F2, and F3 had hardness values of 9.84 kgF, 8.04 kgF, and 7.14 kgF, respectively. The higher the concentration of Eudragit polymer present in these formulations, the greater the hardness. These observations could be due to stronger bonds formed with the hydrophobic polymer (Eudragit). Other researchers reported similar findings when compacts formed with methacrylic polymers (Eudragit L100-55 and Eudragit L100) were compared with that formed with hydroxypropyl methylcellulose (HPMC). Tatavarti et al.26 and Naveen et al.27 observed weaker compact formation with HPMC than with methacrylic polymers.
The friability percentage ranged between 0.5% and 1.04%, except batch F5, with a friability percentage of 1.51%. Thus, most tablets met the pharmacopeia requirement for uncoated tablets. The results showed the ability of tablets to withstand some reasonable levels of abrasion during handling and transportation, except batch F5, which contained hydrophilic polymer (CMC) only.
Floating and swelling properties of tablets
The floating lag time and floating time of the various tablets are shown in Table 4, while the swelling indices are shown in Figure 2. The floating lag time for batches F1 to F5 was within 49 min. Batch F4, prepared by coacervation, floated within 1 min but disintegrated immediately and lost its integrity. This may have been due to insufficient binder (batch F4 had the lowest concentration of CMC). Batch F5, which contained only CMC, had the lowest floating lag time. The results showed variation in floating lag time with different polymer ratios used. Of all the formulations that contained both Eudragit and CMC, batch F1, which contained Eudragit and CMC in 1:2 ratio, had the lowest floating time, while batch F3, with a Eudragit to CMC ratio of 1:1, had the highest floating lag time. The total floating time for batch F3 was 3 h, while batches F1, F2, and F5 floated for more than 8 h.
It was observed from the present study that the floating lag time and total floating time were functions of both the hydrophilic (CMC) and hydrophobic (Eudragit) polymers present. The higher the concentration of hydrophilic polymer, the lower the floating lag time (see batch F5). Moreover, the higher the concentration of hydrophobic polymer, the higher the floating lag time (see batch F3).
Content uniformity
The assay results ranged from 96.82% to 102.12% as shown in Table 3. Controlled release theophylline bilayer floating tablets contained not less than 90.0% and not more than 110.0% of the labeled amount of theophylline.28 From the result obtained (96.82-102.12%) as shown in Table 3, the bilayer floating tablets from all the formulations passed the drug content test. It is important for the tablets to have uniform content of the active ingredients, as this would guarantee the therapeutic effectiveness of all the tablets produced.
In vitro drug release profiles
Figure 3 shows the dissolution profiles of the various batches. Two distinct phases of release were observed in batches F1, F2, F3, and F4: one for the IRL and the other for the controlled release layer. All formulated bilayer tablets showed controlled release of drug over 8 h, while batch F6 (conventional tablets) released the entire drug content within 2 h. The maximum percentage drug release by batches F1, F2, F3, and F5 was 75%, 70%, 80%, and 73%, respectively. Batch F2, which contain Eudragit and CMC in 1:5 ratio, was better prolonged than any other batch (Figure 3). Table 5 illustrates the values of the release rate constants (K) and the regression coefficients (R2) for each model for the six batches of tablets in 0.1 N HCl using a basket at 100 rpm. Research has shown that the model that best fits the release data should be the one with the highest R2 values when analyzed for zero order, first order, and Higuchi models.29 The Higuchi equation was found to have the highest R2; thus release of theophylline from the various matrix tablets is by drug diffusion.
CONCLUSION
Bilayer floating tablets of theophylline were the focus of this research. This is an approach to achieve in vitro immediate release, buoyancy, and prolonged release. The various sets of granules had a good flow property; combination of Eudragit RL100 and CMC produced granules with a better flow property. The presence of gel-forming polymers (CMC and Eudragit RL100) and a gas-producing agent (sodium bicarbonate) helps to achieve prolonged release. Citric acid helps to promote buoyancy under elevated pH of the stomach, thus enhancing drug release. A prolonged floating time and shorter floating lag time could be achieved by appropriate combination of CMC and Eudragit. The ratio of Eudragit and CMC affects the drug release rate and mechanism of release. The in vitro drug release profiles obtained with combination of Eudragit and CMC in 1:2 ratio (F1) produced a prolonged floating duration (>8 h) and a shorter floating lag time (20 min), attributes of a controlled released product. Thus, appropriate combination of hydrophobic and hydrophilic polymers can produce well retarded bilayer floating tablets.
ACKNOWLEDGEMENTS
The authors are grateful to the management of Evonik Industries AG, Werk Röhm, Darmstadt, for the gift of the Eudragit® RL100 sample. Also to be acknowledged is the management of Vital Biotic, Nigeria Limited, for providing the API (theophylline powder) at no cost. Special thanks to the laboratory staff of Pharmaceutics and Industrial Pharmacy Department, Delta State University, Abraka, Nigeria, for the use of the laboratory and for helping to operate some of the machines.
Conflicts of interest: No conflict of interest was declared by the authors. The authors alone are responsible for the content and writing of the paper.