INTRODUCTION
Solid dispersions and cocrystals have emerged in the evolution of supramolecular systems, such as hybrid liquisolid systems, i modern investigations.1,2 Crystal engineering is the process of creating supramolecules with specific structures and distinct physicochemical properties. Cocrystals are a general term for all products that have non-covalent or ionic intermolecular interactions between two or more dissimilar molecules with certain stoichiometric ratios in the crystal lattice and are prepared or developed using a crystal engineering approach;3,4 one of the moieties selected must be an active moiety. Generally, the conformer, which often has no pharmacological efficacy, is chosen from the generally recognized as safe (GRAS) or everything added to food in the United States list.5,6 Therefore, a practical approach is used for organizing the physicochemical properties that have no medical efficacy and diversity.7,8 Over the past few decades, many studies have demonstrated a significant increase in the use of the cocrystallization approach and its feasibility in a formulation as an optimized approach for modulating physicochemical and biopharmaceutical properties of active moieties.9,10
Rosuvastatin calcium (RSC) is a synthetic biopharmaceutical classification system class-II drug that is used to treat hyperlipidemia by increasing high-density lipoprotein cholesterol and decreasing triglycerides, low-density lipoprotein cholesterol, and apolipoprotein. It can be used to decrease the progression of atherosclerosis and prevent coronary heart diseases.11 Rosuvastatin, also known as “super statin”, has demonstrated acceptable results in terms of potency and safety.12,13 RSC exhibits low solubility in gastrointestinal fluids because it has low water solubility (0.33 mg/mL) due to its crystalline nature.14,15 RSC has a 20% oral bioavailability due to pervasive first-pass hepatic biotransformation,16 and high doses have been associated with increased hematuria, proteinuria, serum creatinine, and rhabdomyolysis.17
Cocrystallization of RSC may be a possible formulation path for increasing the bioavailability of molecules without changing the chemical integrity and upholding the physical stability. RSC cocrystals with coformers, such as sorbitol18 and vanillin,19 have been generated with improved bioavailability, resulting in their ability to modify physicochemical properties.20 Ferrari et al.21 reported methods for preparing RSC using three RSC cocrystals, such as rosuvastatin 2-aminopyrimidine hemihydrate, rosuvastatin pyrazine hydrate, and rosuvastatin quinoxaline.
In most cases, the choice of coformer should be based on the Cambridge structural database (CSD), functional/structural possessions, and risk-free GRAS status.22,23 The chosen conformer must contain a group that can develop molecular synthons with active moieties. As an applicable companion, choosing amino acids that fall under the GRAS group is a beneficial option because they are inexpensive and have low toxicity. Researchers have previously reported that amino acids form salt when they react with several classes of therapeutic agents.24,25
This study aimed to use the solvent evaporation method to develop RSC cocrystals with the L-asparagine (ASN) conformer from the GRAS group in order to modify the physicochemical properties of the molecule. Figure 1 depicts the molecular structures of RSC and ASN. The obtained cocrystals were characterized by infrared (IR) spectroscopy, differential scanning calorimetry (DSC), powder X-ray diffraction (PXRD), and scanning electron microscopy (SEM). In vitro studies, such as apparent solubility studies, dissolution studies, and stability studies, were conducted on RSC and its cocrystals, and the results were compared.
MATERIALS AND METHODS
Materials
RSC was obtained from Apex laboratory Pvt ltd., Chennai. ASN and methanol (chromatographic grade) were purchasedfrom Lotus chemicals. The chemicals used in the study were of analytical reagent grade.
Coformer selection
Amino acids may be the initial attraction in the formation of cocrystals. Their zwitterionic potential allows them to form zwitterionic cocrystals.26,27 Based on the CSD,28 structural research, and Scifinder literature scanning program, Tilborg et al.27 provided a list of amino acids that indicate the formation of cocrystals in his mini-review, which may be useful to many researchers in choosing a suitable amino acid as a coformer. According to his findings, ASN does not carry any controversial side chains, allowing it to exist in a zwitterionic state and produce cocrystals.29. The stoichiometric ratio of the drug and coformer was selected for this study because of its safety and effectiveness in the preparation of the cocrystals.
Rosuvastatin-ASN cocrystal synthesis
The stoichiometric ratio of RSC (150 mg) and ASN (42 mg) was solubilized in 5 mL of methanol and 1.5 mL of water separately to form a clear solution using a sonicator. Both solutions were mixed in a magnetic stirrer at 900 rpm. The stirring process continued until the solvent evaporated completely at room temperature. To ensure the removal of the unbound drug and conformer, the obtained cocrystals were washed 3-5 times with methanol/water (5:1) and filtered using 0.45 µm membrane filters. The resulting product was dried overnight in a desiccator. The solvent evaporation method was used to produce large-scale samples for evaluation.
Aqueous solubility studies
Solubility analysis was conducted in distilled water and pH 1.2 buffer, pH 4.5 buffer, and pH 6.8 phosphate buffer solutions. Excess amounts of RSC (approx. 50 mg) and RSC-ASN (RSC-C) cocrystals (Eq. wt. of 50 mg of pure RSC) were placed in screw-capped vials containing 10 mL of distilled water and pH 6.8 phosphate buffer, with continuous stirring for 48 h in a water bath shaker. This was maintained at 37°C and 200 rpm and filtered using a 0.45 µm membrane filter. A quantitative analysis of RSC in the filtrate was performed spectrophotometrically using an ultraviolet (UV) spectrophotometer (V-630, Jasco, Japan) at 244 nm. Equilibrium analysis was conducted three times, and the residual solvents were centrifuged and analyzed by Fourier-transform IR (FTIR) spectroscopy.
In vitro dissolution studies
In vitro dissolution of RSC and the obtained cocrystals was determined using a 900 mL buffer solution (i.e., pH 6.8 phosphate buffer) at 37°C and 50 rpm as per U.S. Pharmacopeia (USP). This was performed using a USP Apparatus-I dissolution vessel (TDL-08L, Electrolab, India). The highest doses of RSC (40 mg) and cocrystals (Eq. wt. of RSC 40 mg) were weighed, and the intact powder was placed in a dissolution vessel. At time intervals of 0, 10, 20, 30, 40, 50, and 60 min, 5 mL of the samples were collected and replenished with a new buffer. The drug content in the filtered sample was measured spectrophotometrically using a UV spectrophotometer (V-630, Jasco, Japan).
PXRD analysis
A PXRD diffractometer (Philips Xpert MPD, Philips, Holland) was used to obtain diffractogram patterns of RSC, ASN, and the obtained product. The instrument was equipped with a Cu target X-ray tube source and a Xe-filled counteract or a proportional detector. The diffraction data were collected by maintaining tube voltage and current at 30 kV and 15 mA, respectively, for 2θ scan axes using a scan range of 5°-65°, a step width of 0.02°, and a scan speed of 10.00°/min. The JCPDF database software was used to determine the peak intensity of each sample.
FTIR spectroscopy
FTIR spectra of all the samples were obtained using the FTIR Azilent carry 360 series, which consists of a DLATGS detector with a 2 cm-1 spectra resolution. A powdered sample of 2-4 mg was kept in a sample holder and scanned over a range of 4000-400 cm-1, and the obtained data were analyzed using the OPUS spectral software.
SEM analysis
Surface images of the pure RSC drug, ASN coformer, and obtained cocrystals were acquired at various magnifications using a SEM (XL30ESEM) with EDAX equipped with a secondary and backscattered electron detector. Samples were attached to carbon tabs, placed on aluminum pin stubs, and sputter-coated with gold/palladium under vacuum in preparation for SEM analysis.
DSC analysis
All the samples were measured using a Mettler Toledo DSC 821e instrument under nitrogen purge (30 mL/min). Powder samples (2-5 mg) were loaded into aluminum pans and sealed. The samples were scanned at temperatures of 30°C to 350°C at a heating rate of 10°C/min. The data was collected using the TAQ series advantage software.
1H liquid fourier-transform nuclear magnetic resonance (FT-NMR) spectroscopy
The pure RSC drug and RSC-C cocrystals were dissolved in deuterated dimethyl sulfoxide, while the ASN conformer was dissolved in deuterated water for FT-NMR analysis. Chemical shifts were observed in the 1H FT-NMR spectra of the RSC, ASN, and RSC-C cocrystals, which were recorded on a 400 MHz FT-NMR spectrometer (model: JNM- ECz 400S).
Product yield and drug content estimation:
The prepared cocrystals were collected and weighed accurately, and the product yield was calculated by dividing the actual weight of the obtained cocrystals by the total weight of the drug and coformer.
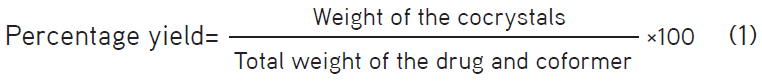
The prepared cocrystals were accurately weighed to be equivalent to 100 mg of the pure drug and dissolved in 100 mL of a pH 6.8 phosphate buffer solution. The solution was filtered, and the drug content was determined using a UV-visible spectrophotometer at 244 nm.
Micromeritic evaluation of the cocrystals
The micromeritic properties of the RSC-C cocrystals were compared to those of the pure RSC drug. The bulk density, tapped density, Hausner ratio, Carr’s index, and repose angle of RSC and RSC-C were all measured. The bulk density was determined using USP method I, whereas the tapped density was determined using USP method II with a tapped density tester (Aymes, Turkey). The following equations were used to calculate the Hausner ratio, Carr’s index, and repose angle.
Hausner ratio = tapped density/bulk density (2)
Carr’s index%= (tapped density – bulk density) × 100/tapped density (3)
Tan θ = 2h/D (4)
Statistical analysis
The results of the percentage yield, micromeritic evaluations, solubility, and dissolution studies were expressed as mean (M) ± standard deviation.
RESULTS
Aqueous solubility analysis
Solubility analysis was conducted in distilled water and pH 1.2 buffer, pH 4.5 buffer, and pH 6.8 phosphate buffer solutions. The pure RSC concentrations in water, pH 1.2 buffer, pH 4.5 buffer, and pH 6.8 phosphate buffer were 0.836 mg/mL, 0.624 mg/mL, 1.143 mg/mL, and 1.427 mg/mL, respectively, after 48 h of continuous stirring. The solubility of the physical RSC-C mixture did not differ significantly from that of the pure active pharmaceutical ingredient (API). When compared to pure RSC, the RSC-C cocrystals exhibited a 2.17-fold, 2.21-fold, 2.29-fold, and 2.42-fold increase in the solubility in water, pH 1.2 buffer, pH 4.5 buffer, and pH 6.8 phosphate buffer, respectively (Table 1). The crystal arrangement and cocrystallization process could be the reasons for these outcomes.30 Since RSC is poorly soluble in water,14,15 the cocrystal solubility greatly relies on the solubility of its composition.31 When compared to that of pure RSC, the improved solubility could also be due to its relatively small particle size. This establishes the possibility of RSC-C cocrystals to be used in new product formulations.
PXRD analysis
The diffractogram pattern of the RSC-C cocrystals exhibited unique crystalline peaks when compared to those of RSC and the coformer. Figure 2 shows the PXRD pattern of the pure drug, coformer, and obtained cocrystals.
The pure RSC showed a single 2θ scattering angle at 43.25°, indicating the amorphous nature of the drug.32 The RSC-C cocrystals showed new characteristic peaks at 9.14°, 10.28°, 20.85°, 24.61°, 32.93°, and 40.06°, which were absent in the pure RSC and ASN diffractogram patterns. However, in the cocrystals, the ASN peaks of 11.85°, 17.70°, 18.23°, 19.91°, 27.84°, and 43.15° shifted to 11.71°, 17.53°, 18.05°, 19.73°, 27.72°, and 42.46°, respectively. When comparing the diffractogram pattern of the cocrystals to that of the pure drug and conformer, the appearance/disappearance and shifting of the peaks in the cocrystal diffractogram pattern indicate the evaluation of a new crystalline phase.
FTIR studies
FTIR spectroscopy was used to demonstrate the interaction between RSC and ASN. The FTIR spectra of RSC showed characteristic peaks corresponding to carboxylic O-H stretch at 3382 cm-1, N-H stretch at 2968 cm-1, C=C stretch at 1541 cm-1, asymmetric vibration of CH3 at 1436 cm-1, C-F stretch at 1149 cm-1, symmetric vibration of CH3 at 1379 cm-1, and C-H plane bending of the aromatic ring at 775 cm-1.33 ASN had characteristic peaks corresponding to O-H stretch at 3436 cm-1, N-H stretch at 2924 cm-1, and C=O stretch at 1716-1750 cm-1.34
Figure 3 shows the IR spectra of RSC, ASN, and the RSC-C cocrystals. When compared to those of the pure RSC, the N-H group of the cocrystals shifts to 2931 cm-1, while the O-H group shifts to 3421 cm-1, indicating new hydrogen bond formation in the cocrystals. Few additional peaks in the range of 1638-1750 cm-1 were observed in addition to the characteristic peaks of RSC because of the carboxylic acid moiety, which expresses the presence of C=O stretching in the cocrystals. When compared to the pure RSC, the cocrystals exhibited a change in the chemical environment, which was consistent with the PXRD and DSC results and confirmed the interaction between RSC and ASN.
SEM analysis
Figure 4 shows the SEM micrographs of RSC, ASN, and the RSC-C cocrystals obtained by solvent evaporation. RSC had irregular granular-shaped particles, with not much difference in the morphology of the pure RSC and physical RSC-C mixture. ASN exhibited a stick-shaped crystalline morphology. The cocrystals obtained via solvent evaporation underwent a phenomenal transformation into an irregular closely fitted crystalline structure. The formation of intermolecular hydrogen bonds between RSC and ASN could be the cause of the change.
DSC analysis
DSC data of the RSC-C cocrystals were obtained and compared with those of the pure drug and coformer, and the results are shown in Figure 5. The DSC thermogram of the pure RSC drug showed broad endothermic peaks at 80.7°C and 217.9°C, indicating that the drug substance had an amorphous form.35 The thermal curve of ASN exhibited a sharp endothermic peak at 116.1°C and 237.1°C with high enthalpy. The physical mixture (1:1 ratio) of RSC and ASN exhibited endothermic peaks at 101.8°C and 237.1°C, with slight shifting and broadening of peaks. This may be due to the loss of purity of each compound when mixed together rather than a fundamental indication of incompatibility. The RSC-C cocrystal obtained by solvent evaporation had a unique characteristic sharp endothermic peak at 127°C with a high intensity that was different from those of the pure RSC and ASN coformer. According to many researchers, most cocrystals melt at a temperature that is different from that of their APIs and coformers. According to the Perlovich36 study, the melting points of the obtained cocrystals are often in the middle of (55.3%), less than (38.9%), or higher than (14.5%) that of the starting materials. The RSC-C cocrystals exhibited middle endotherm when compared to the starting materials on the DSC profile, which is a very common phenomenon in 1:1 stoichiometric cocrystals. The changes in the crystalline structure and cocrystal formation are indicated by a change in the melting point. The PXRD and FTIR analyses confirmed these changes.
1H liquid FT-NMR spectroscopy
Figure 6 shows the 1H FT-NMR spectra of RSC, ASN, and the RSC-C cocrystals. When the NMR spectra of the RSC-C cocrystals were compared with the pure drug (RSC) spectra, changes in the chemical shift values were observed at 3H from 3.395 to 3.428 and 6H from 3.500 to 3.533, respectively. The chemical shift value confirmed cocrystal formation between the drug and conformer, which was probably due to interactions between the free hydroxyl group of RSC and the amine moiety of ASN. Furthermore, the FT-NMR analysis results were in total agreement with the PXRD, DSC, and FTIR analysis results.
In vitro dissolution analysis
The cumulative dissolution rate was determined for the pure RSC and RSC-C cocrystals obtained by solvent evaporation. The dissolution rate was measured in pH 6.8 buffer, and the results are presented in Figure 7. RSN showed a 0.86% dissolution rate immediately after the addition of a solvent, whereas RSC-C cocrystal presented 3.8%. After 10 min, the cocrystals had a dissolution rate of 23.23%, while the pure drug had a 9.42% dissolution rate. The dissolution rate of the Pure RSC increased slowly and reached 42.64% within 2 h, whereas that of the cocrystals increased quickly, reaching 91.02% within 2 h. The increase in the dissolution rate of the RSC-C cocrystals is because of the change in the crystal morphology and the tiny particle size, as shown in the SEM analysis.37 A change in the melting point of the DSC profile indicates more dissolution.38,39 The FTIR, PXRD, SEM, DSC, solubility, and dissolution analyses revealed altered dissimilarities between the cocrystal properties and the pure API properties, which are due to the change in the crystalline structure.
Percentage yield and drug content estimation
The percentage yield and drug content estimation of the obtained cocrystals were conducted in triplicate. The percentage yield of the cocrystals was found to be 95.75%±0.25%, while that of the drug content was estimated to be 98.4%±0.43%. The results indicated that the obtained cocrystals had a good product yield with an acceptable drug content.
Micromeritic evaluation of the cocrystals
Table 2 shows the micromeritic properties of RSC and RSC-C. The repose angle of the RSC-C cocrystal was 29°.81’±0.45, whereas that of the pure RSC was 32°.31’±0.52. In comparison to the pure RSC, this shows that the cocrystals have good flow properties. The compressibility of the RSC-C cocrystals was found to be 6.32%±0.68%, whereas that of the pure RSC was 11.24%±0.46%. The result demonstrated in comparison to the pure RSC, the cocrystals have excellent compressibility. The Hausner ratios of RSC-C and RSC were found to be 1.06%±0.02% and 1.18%±0.21%, respectively, indicating that RSC-C had better flow properties and compression strength than RSC.
DISCUSSION
The functional groups present in the drug and coformer molecules were the primary determinants of cocrystal formation in the synthon method40. The RSC consists of two oxydrillic groups that are bound to asymmetric carbon atoms. Therefore, ASN, which contains an amide group, was chosen as a coformer. The structural characterization confirmed the expected structural modification. The SEM analysis results revealed that the irregular granular nature of pure RSC changed to a closely fitted crystalline structure in the cocrystals. The average particle size of the RSC-C cocrystals was 4.3 µm, which is in between the average particle size of pure RSC and ASN. The alteration in the crystalline habit and the average particle size may have influenced the formation of a hydrogen bond between the drug and conformer. In the FTIR analysis, alterations in chemical structure included the broadening of the O-H stretching peak at 3382 cm-1 in the RSC-C cocrystals when compared to RSC, confirming the hydrogen bond interaction. The broad endothermic peak of RSC changed to a sharp endothermic peak in the RSC-C cocrystals with different melting points, indicating partial crystallization. According to Perlovich’s41 investigation, 28.9% of the cocrystal formulation had a lower melting point than the parent molecules. The decrease in the melting point and enthalpy indicates the formation of a weak crystalline structure.42,43,44 According to the PXRD diffractogram, RSC had an amorphous form. This was confirmed by the broad peaks in the DSC profile.45 However, new characteristic peaks other than RSC and ASN peaks were observed in the RSC-C cocrystals, indicating a change in the chemical environment and transition from a semi-crystalline form to a crystalline form. The 1H liquid FT-NMR spectra revealed an exact shift in two oxydrillic groups to an up-field nature, indicating the transfer of electrons from the hydroxyl group during hydrogen bond interaction. All these results confirm the formation of cocrystals between the drug and coformer because of interactions between the free hydroxyl group of the drug and the amine group of the coformer. Afterward, comparative studies on the solubility and dissolution rate were conducted, and the results showed that the cocrystals had almost 2-fold higher solubility and dissolution rates than the parent molecule. The cocrystals demonstrated better micromeritic properties (flow properties, compressibility, and compression strength) than RSC, providing them an advantage in pelleting and tableting properties.
CONCLUSION
RSC-C cocrystals were successfully prepared using the solvent evaporation cocrystallization technique. The solubility and dissolution rates of the obtained cocrystals were almost 2-fold higher than those of the parent molecule. This demonstrates the removal of impediments to RSC entering the bloodstream, such as low solubility and low dissolution rates. The formation of a new crystalline form was affirmed by FTIR, PXRD, and FT-NMR analyses. These results were supported by a change in the melting point in the DSC thermogram compared to that of the pure RSC and ASN. The enhanced physicochemical properties of RSC contribute to the viability of novel formulations. According to the results, the micromeritic properties of the cocrystals provide the advantages of pelleting and tableting properties. The strategy for transforming a cocrystal idea into an application has been effectively explained in the literature, which is mainly helpful for the development of the process in future investigations.46,47,48 However, the in vivo performance of RSC cocrystals, such as AUC and Cmax, was not covered in this study, but it will be included in the next study. The results clearly indicate that this cocrystallization method improved the dissolution and solubility of RSC and caused a change in its chemical structure.
ACKNOWLEDGMENTS
The authors are thankful to Maharajah’s College of Pharmacy, Vizianagaram, Andhra Pradesh and GITAM Deemed to be University, Vsakhapatnam for providing the facilities to carry the research work.
Conflict of interest: No conflict of interest was declared by the authors. The authors are solely responsible for the content and writing of this paper.